Investigation of OOS Results in Analytical Testing
Investigation of OOS Results in Analytical Testing
1. Definition
An Out of Specification (OOS) result occurs when an analytical test result for a product, raw material, or intermediate falls outside the approved acceptance criteria or specifications set by regulatory filings or SOPs.
GMP guidelines (e.g., US FDA, MHRA, WHO) require that all OOS results be thoroughly investigated to determine their cause before any batch disposition decision is made.
2. Regulatory Expectation
-
21 CFR 211.192: All drug product production and control records must be reviewed, and any unexplained discrepancy must be thoroughly investigated.
-
Investigations must be timely, documented, and scientifically sound.
3. OOS Investigation Process
Step 1: Immediate Actions
-
Notify QA and stop further processing/testing of the batch.
-
Quarantine affected material/batch.
-
Review raw data immediately to check for obvious errors.
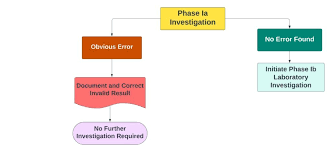
Step 2: Phase I – Laboratory Investigation
-
Objective: Identify any laboratory errors.
-
Activities include:
-
Review of analytical raw data, chromatograms, and calculations.
-
Verification of instrument calibration, system suitability, and environmental conditions.
-
Check reference standards, reagents, and sample integrity.
-
Interview analysts involved.
-
-
If a clear analyst or laboratory error is confirmed → Retest as per SOP.
Step 3: Phase II – Full-Scale Investigation
-
Triggered when no lab error is found.
-
Evaluate potential manufacturing or process-related causes:
-
Review batch manufacturing records, equipment logs, deviations, and environmental monitoring data.
-
Assess material quality, storage, and handling.
-
Check for any changes in process parameters.
-
-
Determine if OOS is due to:
-
True OOS (product genuinely out of spec)
-
Assignable cause (identified and explained)
-
Inconclusive
-
Step 4: Hypothesis Testing / Retesting
-
Retesting must be scientifically justified and approved by QA.
-
Use newly prepared samples from the same retained batch unit.
-
Avoid “testing into compliance.”
Step 5: Conclusion & Documentation
-
Investigation report must include:
-
Description of OOS
-
All actions taken and timelines
-
Root cause determination
-
Risk assessment
-
Corrective & Preventive Actions (CAPA)
-
-
QA review and approval are mandatory before batch disposition.
4. Key GMP Points
-
Never discard initial failing data.
-
All OOS results must be reported, even if subsequent retesting passes.
-
Follow ALCOA+ principles for data integrity.
-
Train analysts regularly on OOS handling procedures.
-
Trend OOS data for continuous improvement.
5. Common Causes of OOS in Analytical Testing
-
Analyst error (weighing, pipetting, dilution mistakes)
-
Equipment malfunction or miscalibration
-
Degraded or contaminated samples
-
Manufacturing process deviations
-
Raw material variability
✅ Summary Flow:
Detection → Lab Investigation → Full-Scale Investigation → Root Cause → CAPA → QA Review → Batch Disposition → Trending
🎓 Discover one of the best Pharmaceutical Quality Assurance course available —click below to explore the course that’s shaping future Quality Assurance skills.
https://trcjw.on-app.in/app/oc/306166/trcjw